Страница: 4/5
k = а* К*,
одна из которых a* - константа мономолекулярного превращения активированного комплекса в продукты реакции, имеющая размерность частоты, а вторая К* - константа равновесия образования переходного комплекса.
В общем случае реакция может идти от исходных веществ к конечным продуктам различными путями, т.е. через различные перевалы(величины энергии активации). Однако, реакция, как правило, идет по одному из путей, такому, где энергетические затраты будут наименьшими.
Ускорение химических реакций с помощью кислот и оснований - наиболее распространенный
прием из используемых химиками в повседневной работе. Мы рассмотрим только катализ
«протонными» кислотами. В этом случае каталитически действующим началом является
ион гидроксония ![]() образующийся
при диссоциации кислоты в водных растворах
образующийся
при диссоциации кислоты в водных растворах
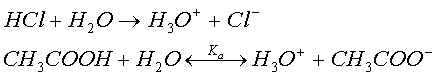 (б)
(б)
При умеренной концентрации соляная кислота полностью распадается на ионы. Карбоновые кислоты, в частности уксусная (б), диссоциируют не полностью: устанавливается определенное равновесие между ионами и недиссоциированными формами кислоты. В качестве меры диссоциации слабых кислот выбирают константу диссоциации, которая для уксусной кислоты равна 1,75*10^-15 моль/литр;
В чистом виде протон H+ в растворе не существует, так как ему выгоднее соединиться с молекулой воды. (Однако для краткости записывают просто H+, подразумевая под этим символом ион гидроксония.)
удобно выражать концентрацию водородных ионов в единицах рН=-lg[H+], т. е. в единицах показателя степени (эту единицу измерения впервые ввел Сёренсен).
Чем выше концентрация кислоты (или кислотность среды), тем больше скорость реакции, но лишь до определенного значения pH. Исходя из этого попробуем разобраться в двух вопросах:
1) почему растет скорость с увеличением концентрации Н+ионов (т. е. с уменьшением рН);
2) почему реакция замедляется при добавке кислоты сверх определенной нормы;
Существует твердая уверенность в том, что все начинается с атаки атома азота на углерод карбонильной группы. Азот располагает двумя неспаренными электронами, а углерод не только их не имеет, но даже обладает некоторым дефицитом электронной плотности. Говорят, что карбонильная группа поляризована - часть внешнего электронного облака смещена в сторону кислородного атома.
Когда мы вводим в реакционную смесь кислоту, то образовавшиеся водородные ионы начинают атаковать молекулы обоих партнеров, но только один вид атаки будет способствовать химическому взаимодействию их - атака на карбонильный кислород. Почему же именно так ? Да потому, что координация протона с атомом кислорода приведет к смещению электронной плотности от атома углерода в сторону протона. Произойдет оголение углерода, и он сможет легко принять электроны атома азота. В этом, собственно, и заключена природа кислотного катализа. Нетрудно сообразить, что чем больше концентрация H+ ионов (повторяем - чем меньше рН), тем больше концентрация протонизованных (по кислороду) молекул альдегида, тем выше должна быть скорость реакции.
Мы рассмотрели, конечно, упрощенную схему кислотного катализа, но она является хорошей иллюстрацией того, как изучают явление и к каким выводам можно прийти в результате знания зависимости скорости реакции от концентрации водородных ионов. Анализ явлений катализа под действием ионов ОН- (основной катализ) принципиально мало отличается от только что рассмотренного анализа кислотного катализа.
При изучении катализа органических реакций в сильно кислых средах встречаются с трудностями, которые обычно легко преодолеваются, когда работают с разбавленными кислотами. Но не будем заострять на этом внимание, обратим его лишь на то, какого рода информацию получают, изучая концентрационные зависимости.
Гомогенный катализ
Среди многочисленных каталитических реакций особое место занимает катализ в цепных реакциях.
«Цепными реакциями, как известно, называются такие химические и физические процессы, в которых образование в веществе или в смеси веществ некоторых активных частиц (активных центров) приводит к тому, что каждая из активных частиц вызывает целый ряд (цепь) последовательных превращений вещества» (Эмануэль, 1957).
Такой механизм развития процесса возможен благодаря тому, что активная частица взаимодействует с веществом, образуя не только продукты реакции, но и новую активную частицу (одну, две или более), способную к новой реакции превращения вещества, и т. д. Возникающая при этом цепь превращений вещества продолжается до тех пор, пока активная частица не исчезает из системы (происходит «гибель» активной частицы и обрыв цепи). Наиболее трудная стадия при этом - зарождение активных частиц (например, свободных радикалов), после же зарождения цепь превращений осуществляется легко.
Цепные реакции широко распространены в природе. Полимеризация, хлорирование, окисление и многие другие химические процессы идут по цепному, а точнее - по радикально-цепному (с участием радикалов) механизму.
Механизм окисления органических соединений (на ранних стадрях) в настоящее время установлен достаточно тщательно. Если обозначить окисляющееся вещество R-H (где Н - атом водорода, имеющий наименьшую прочность связи с остальной молекулой R), то этот механизм можно записать в следующем виде:
Катализаторы, например соединения металлов переменной валентности, могут оказывать влияние на любую из рассмотренных стадий процесса.
Остановимся теперь на роли катализаторов в процессах вырожденного разветвления цепей. Взаимодействие гидроперекиси с металлом может приводить как к ускорению так и к торможению реакции окисления органических веществ соединениями металлов переменной валентности в зависимости от характера продуктов, образующихся при распаде гидроперекиси. Соединения металлов образуют с гидроперекисями комплекс, который распадается в «клетке» растворителя среды, если обра-зующиеся при распаде комплекса радикалы успеют выйти из клетки, то они инициируют процесс (положительный катализ). Если же эти радикалы не успеют выйти и рекомбинируют в клетке в молекулярные неактивные продукты, то это приведет к замедлению радикально-цепного процесса (отрицательный катализ), поскольку в этом случае гидроперекись - потенциальный поставщик новых радикалов- расходуется вхолостую.
До сих пор мы рассматривали лишь неглубокие стадии процессов окисления; на более глубоких стадиях например в случае окисления углеводородов, образуются кислоты, спирты, кетоны, альдегиды, которые также могут реагировать с катализатором и служить дополнительным источником свободных радикалов в реакции, т. е. в этом случае будет налицо дополнительное вырожденное разветвление цепей.
Гетерогенный катализ
К сожалению, до сих пор, несмотря на достаточно большое число теорий и гипотез в области катализа, многие основополагающие открытия были сделаны случайно или в результате простого эмпирического подхода. Как известно, случайно был найден ртутный катализатор сульфирования ароматических углеводородов М. А. Ильинским, который нечаянно разбил ртутный термометр: ртуть попала в реактор, и реакция пошла. Аналогичным образом были обнаружены теперь всем хорошо известные, а в свое время открывшие новую эру в процессе полимеризации катализаторы стереоспецифической полимеризации Циглера.
Естественно, что такой путь развития учения о катализе не соответствует современному уровню науки, и именно этим объясняется повышенный интерес к изучению элементарных стадий процессов в гетерогенно-каталитических реакциях. Эти исследования - прелюдия для создания строго научных основ подбора высокоэффективных катализаторов.
Во многих случаях роль гетерогенных катализаторов в процессе окисления сводится
к адсорбции органического соединения и кислорода с образованием на поверхности
катализатора адсорбированного комплекса этих веществ. Такой комплекс разрыхляет
связи компонентов и делает их более реакционноспособными. В некоторых случаях
катализатор адсорбирует лишь один компонент, который диссоциирует на радикалы.
Например, пропилен на закиси меди диссоциирует с образованием аллильного радикала
![]() , легко вступающего
затем в реакцию с кислородом.
, легко вступающего
затем в реакцию с кислородом.
Выяснилось, что каталитическая активность металлов переменной валентности в значительной мере зависит от заполнения d-орбиталей в катионах окислов металлов.
По каталитической- активности в реакции разложения многих гидроперекисей соединения металлов располагаются следующим ря-
Мы рассмотрели один из возмжных путей инициирования процесса - взаимодействие гидроперекиси с катализатором. Однако в случае окисления реакция гетерогенного инциирования цепей может протекать как путем распада на радикалы гидроперекиси, так и путем взаимодействия углеводорода с кислородом, активированным поверхностью катализатора. Инициирование цепей может быть обусловлено участием заряженной формы органического соединения RH+, образующегося при взаимодействии RH с катализатором. Так обстоит дело с катализом в реакциях инициирования (зарождения и разветвления) цепей. Роль гетерогенных катализаторов в реакциях продолжения цепи особенно четко подчеркивается изменением скорости и направления изомеризации перекисных радикалов.
Катализ в биохимии
Реферат опубликован: 15/04/2005 (9805 прочтено)