Страница: 5/7
![]() (35)
(35)
Следует отметить, что уравнение (35) является лишь обобщением формы уравнения адсорбции Гиббса и физически совершенно эквивалентно уравнению (34). Более того, можно сказать, что, уступая уравнению (34) в простоте, уравнение (35) и усложняет интерпретацию величины s, поскольку утрачивается аналогия с натяжением упругой мембраны. Строго говоря, термин «поверхностное натяжение» применим только к поверхности натяжения.
Другой обобщенной и также физически эквивалентной формой является запись уравнения адсорбции Гиббса для слоя конечной толщины [24]
Ads=![]() (36)
(36)
где Va и Vb — части объема Vs поверхностного слоя, разделенные поверхностью натяжения.
В случае плоской поверхности уравнение (17) принимает вид [4, 17, 18]
![]() (37)
(37)
и соответствует уравнению (32).
Выше мы указывали, что уравнение (34) было получено Гиббсом для границы флюидных фаз. Соответствующее уравнение для плоской твердой поверхности в изотропном состоянии было выведено Эрикссоном [30]
![]() (38)
(38)
где
g — механический аналог поверхностного натяжения жидкости (истинное поверхностное натяжение твердого тела);
s — термодинамический аналог поверхностного натяжения жидкости (условное поверхностное натяжение твердого тела).
В общем случае анизотропной поверхности твердого тела уравнение адсорбции принимает вид [26, 27]
![]() :
:
![]() (39)
(39)
где
![]() — тензор избыточных
поверхностных напряжений;
— тензор избыточных
поверхностных напряжений;
![]() — единичный тензор;
— единичный тензор;
![]() — тензор поверхностной
деформации; символ : означает скалярное произведение тензоров.
— тензор поверхностной
деформации; символ : означает скалярное произведение тензоров.
В уравнении (39) суммирование производится по всем подвижным компонентам.
Что касается неподвижных компонентов, образующих решетку твердого тела, то
их химические потенциалы не фигурируют в уравнении (39). Гиббс вообще не вводил
понятия химический потенциал неподвижного компонента. Его можно определить
лишь условно и отдельно для каждого направления разреза твердого тела как химический
потенциал в равновесной флюидной фазе, контактирующей с твердым телом по данному
разрезу. Определенный таким образом химический потенциал неподвижного компонента
mi' зависит в каждой точке тела от направления нормали
![]() к мысленной поверхности
разреза.
к мысленной поверхности
разреза.
Кроме того, даже в состоянии истинного равновесия величина mi не будет одинаковой
для всех точек разреза и поэтому при переходе к избыточным величинам для межфазной
поверхности приходится брать избыток от произведения химического потенциала
на массу неподвижного компонента. Для каждого направления
![]() на межфазной поверхности
можно определить величину
на межфазной поверхности
можно определить величину
![]() (40)
(40)
причем существует соотношение [31, 32]
![]()
![]() (41)
(41)
где gn — натяжение на поверхности в направлении
![]() .
.
Подстановка (41) в (39) приводит к уравнению [31, 32]
![]() :
:![]() (42)
(42)
которое также является обобщением уравнения адсорбции Гиббса на случай твердой
поверхности, но сформулировано в терминах избыточного поверхностного напряжения.
Для жидкой поверхности
![]() , и уравнения (39)
и (42) переходят в уравнение адсорбции Гиббса.
, и уравнения (39)
и (42) переходят в уравнение адсорбции Гиббса.
При применении уравнения адсорбции Гиббса к поверхности жидкого электрода в нем появляется дополнительный член, связанный с изменением электрического потенциала. Можно сказать, что для изотермо-изобарических условий этот член был получен самим Гиббсом, поскольку он дал термодинамический вывод уравнения Липпмана. В дальнейшем этот вопрос многократно обсуждался при исследовании электрокапиллярных явлений (см., например, [33 – 35]). Строгий вывод уравнения адсорбции Гиббса для плоского поверхностного слоя электрода был дан Парсонсом [36]. Соответствующую теорию для искривленного слоя можно найти в [25, 14].
К весьма сложным разделам термодинамики поверхностных явлений относится анализ искривленных поверхностей во внешних полях. Гиббсом было начато рассмотрение поверхностных явлений в гравитационном поле. Что касается электрического поля, то результаты были получены значительно позднее. Трудность рассмотрения здесь сильно зависит от того, являются ли соприкасающиеся фазы проводниками или диэлектриками, Задача для соприкасающихся проводников решается сравнительно просто [37], для диэлектриков — значительно сложнее [38].
Важным моментом в развитии термодинамики поверхностных явлений было обобщение уравнения адсорбции Гиббса на случай отсутствия адсорбционного равновесия. Здесь нужно отметить прежде всего работы Дефэя [39, 40], в которых было введено понятие вторичных химических потенциалов ei отражающих зависимость поверхностного натяжения от состояния объемных фаз a и b :
![]() (43)
(43)
В уравнении (43) предполагается, что термическое и механическое равновесие установилось, а диффузионное еще не достигнуто.
Процесс установления адсорбционного равновесия включает трансляционное и вращательное движение молекул, в частности, ориентацию несферических молекул в поверхностном слое. Если ориентация происходит гораздо медленнее трансляционно-диффузионного процесса, то можно представить случай, когда вся неравновесность системы обусловлена процессом ориентации молекул (например, диполей) в поверхностном слое. Для такого случая было предложено обобщение уравнения Гиббса [37]
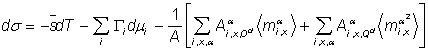 (44)
(44)
где
![]() — среднее значение
составляющей по оси x дипольного момента молекул i‑го компонента в фазе a;
— среднее значение
составляющей по оси x дипольного момента молекул i‑го компонента в фазе a;
![]() — среднее значение
квадрата той же величины;
— среднее значение
квадрата той же величины;
![]() и
и
![]() — соответствующие
сродства; суммирование по x и a означает суммирование по всем составляющим
дипольного момента и по всем фазам и тонким элементарным слоям внутри поверхностного
слоя, рассматриваемым как однородные области.
— соответствующие
сродства; суммирование по x и a означает суммирование по всем составляющим
дипольного момента и по всем фазам и тонким элементарным слоям внутри поверхностного
слоя, рассматриваемым как однородные области.
Следует отметить, что в основе вывода уравнения (44) лежит весьма условное предположение о независимости трансляционных и вращательных составляющих адсорбционно-диффузионного процесса.
Развитие новых направлений в
термодинамике поверхностных явлений
Термодинамика тонких пленок
Гиббс в теории капиллярности ограничился рассмотрением только толстых пленок, в которых можно пренебречь взаимовлиянием поверхностных слоев на противоположных сторонах пленки. Тонкая пленка принципиально отличается от толстой тем что ее поверхностные слои нельзя рассматривать независимо друг от друга. Фактически в тонкой пленке уже нельзя выделить объемную фазу и окружающие ее поверхностные слои, а необходимо рассматривать пленку в целом. Важной характеристикой, отличающей тонкую пленку от толстой, является расклинивающее давление; в опытах оно проявляется в том, что при переходе от толстой к тонкой пленке требуется изменение внешнего давления. Понятие расклинивающего давления было введено Дерягиным [42], которому принадлежат и первые измерения этой величины.
Существует несколько эквивалентных определений расклинивающего давления плоской тонкой пленки. Прежде всего расклинивающее давление П можно определить как разность между значениями внешнего давления Pa на тонкую и толстую пленку
![]() (45)
(45)
где h — толщина тонкой пленки.
Если тонкая пленка образовалась из фазы g и продолжает находиться с ней в равновесии (например, при прилипании пузырька к твердой поверхности: фаза a — газ, фаза g — жидкость), то расклинивающее давление можно определить как:
![]() (46)
(46)
Наконец, поскольку для плоской пленки внешнее давление всегда равно нормальной составляющей тензора давления внутри пленки, можно дать определение
![]() (47)
(47)
и сформулировать его следующим образом:
расклинивающее давление есть разность между нормальным давлением внутри пленки (или внешним давлением) и давлением в объемной фазе той же природы при тех же значениях температуры и химических потенциалов, что и в пленке.
Определение (46) впервые использовали в экспериментальных исследованиях расклинивающего давления [42 – 45], а определение (47) — для расчетов [46].
Реферат опубликован: 15/04/2005 (15923 прочтено)