Страница: 4/8
Рис. 7. Гипотетическая схема эволюции структурной организации локусов генов L-цепей ИГ позвоночных. (Anderson et al., 1995; Gilbert, 1990).
![]()
![]() Обозначения: - сигнальные олигомеры, фланкирующие V и J сегменты.
Обозначения: - сигнальные олигомеры, фланкирующие V и J сегменты.
резкого сокращения количества C генов. В-третьих, подобная организация позволяет упростить регуляцию экспрессии различных локусов.
Ключевым принципом функционирования иммунной системы является принцип клональной селекции иммунокомпетентных клеток, несущих рецептор определенной специфичности. Ряд косвенных данных позволяет считать, что у акул гуморальный иммунный ответ клонально ограничен (Shankey and Clem, 1980). Однако неизвестно, каким образом и насколько эффективно осуществляется это ограничение (Flajnik, 1996). У млекопитающих принцип “одна клетка - одно антитело” обеспечивается благодаря изотипическому и аллельному исключению. При сегментарной организации генов продуктивная перестройка в одном из локусов ведет к блокированию перестройки других локусов и, тем самым к их инактивации. Однако, в случае наличия множественных VJC кластеров, в части из которых VJ сегменты слиты в зародышевой ДНК, механизмы контроля выборочной экспрессии могут быть более сложными или не столь эффективными. Таким образом, переход от кластерной к сегментарной форме организации генов ИГ мог иметь принципиальное значение для формирования полноценной системы гуморального иммунитета.
Вопрос о том, на каком этапе филогенеза произошел этот переход остается открытым. Уже у амфибий гены ИГ L- и Н-цепей организованы сегментарно. При этом, в случае L-цепей наблюдается два основных типа организации: каппа-подобный (один С ген, семейство J сегментов и семейство V сегментов) и лямбда подобный (несколько пар JC и семейство V сегментов). По всей видимости, различные типы L-цепей амфибий произошли от различных типов L-цепей хрящевых рыб, однако относительно невысокая гомология по первичной структуре не позволяет утверждать этого с определенностью (Gilbert, 1990).
У сомика и радужной форели, представителей костистых рыб, организация генов L-цепей и Н-цепей отличается. Гены Н-цепей костистых рыб организованы подобно млекопитающим сегментарно (Bengten et al., 1991). Организация генов L-цепей имеет кластерный характер. В то же время, в отличие от хрящевых рыб, транскрипционная ориентация V генов у них изменена.
Следует отметить, что сомовые и лососевые являются довольно специализированной группой, возникшей в эволюции относительно недавно (Рис. 8). Особенности организации генов легких цепей у этих таксонов могут представлять собой новоприобретенные признаки (Romer, 1966).
В связи с этим особый интерес представляет изучение структры и организации генов L-цепей ИГ у костно-хрящевых рыб. Костно-хрящевые являются архаичной группой костистых рыб, возникшей значительно раньше настоящих костистых. Несмотря на то, что существуют различные мнения относительно последовательности появления костно-хрящевых и двоякодышащих (предков наземных позвоночных) (Ромер и Парсонс, 1992; Юдкин И. И., 1941), очевидно, что костно-хрящевые в большей степени чем коститстые сохранили черты древних первично-костных рыб и, соответственно, с большим основанием могут рассматриваться как промежуточное звено между хрящевыми рыбами и амфибиями.
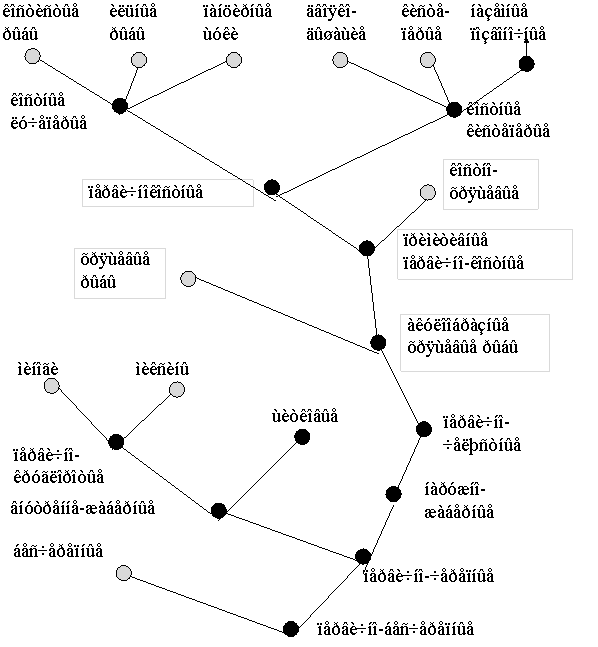
Рис. 8. Эволюционное древо низших позвоночных ( по Северцеву А. Н., 1939).
![]()
![]() Обозначения: - таксоны, имеющие ныне живущих представителей, - таксоны, существование
которых доказано палеонтологическими исследованиями.
Обозначения: - таксоны, имеющие ныне живущих представителей, - таксоны, существование
которых доказано палеонтологическими исследованиями.
МАТЕРИАЛЫ И МЕТОДЫ
ЭЛЕКТРОФОРЕЗ ДНК
В агарозном геле.
Электрофорез ДНК проводили в 1% агарозном геле в 1-кратном буфере ТАЕ, имеющем состав: 0.04 М трис-ацетат, 2 мМ Na2ЭДТА (Маниатис и др., 1984).
В полиакриламидном геле.
Электрофорез ДНК проводили в 6% полиакриламидном геле в 0,5-кратном буфере ТБЕ, имеющем состав: 0,089 М трис-борат, 0,089 М борная кислота, 2 мМ Na2ЭДТА. Для полимеризации геля к 50 мл раствора добавляли 300 мкл 10% раствора персульфата аммония и 30 мкл ТЕМЕД (Маниатис и др., 1984).
ВЫДЕЛЕНИЕ ДНК ИЗ ГЕЛЯ
Адсорбция на диэтиламиноэтилцеллюлозе.
Гель окрашивали в растворе бромистого этидия, участок геля с полосой ДНК вырезали, помещали в камеру и переносили ДНК на диэтиламиноэтилцеллюлозу в 0,5-кратном буфере ТБЕ (см. Электрофорез ДНК (2)) при напряжении 40 В/см. Элюцию ДНК с диэтиламиноэтилцеллюлозы проводили путем инкубирования в течение 30 мин при 70оС в буфере, имеющем состав: 2 мМ NaCl, 1 мМ Na2ЭДТА, 40 мМ трис рН 8,0 (Маниатис и др., 1984).
Адсорбция на кремниевой пудре.
Агарозный гель окрашивали в растворе бромистого зтидия, вырезали участок геля с ДНК и расплавляли инкубированием с двойным объемом 6 М раствора KI в течение 10 мин при 55оС. Затем добавляли суспензию кремниевой пудры (силики) в 3 М растворе KI в концентрации 100 мг/мл из расчета 300 мкг силики на 1 мкг ДНК и инкубировали в течение 2 мин при 55оС. После этого осаждали силику центрифугированием при 2000 g в течение 2 мин и дважды промывали суспендированием в растворе, содержащем 50 мМ NaCl, 10 мМ трис-HCl, рН 8,0, 2,5 мМ Na2ЭДТА и 50% этиловый спирт. Элюировали ДНК с силики суспендированием в воде (Boyle, Lew, 1995).
Замораживание - оттаивание.
Агарозный гель с ДНК 10 мин инкубировали в жидком азоте и осаждали центрифугированием при 12000 g в течение 15 мин, после чего добавляли к супернатанту 1/10 объема 3 М NaAc, 2 объема этанола и осаждали ДНК при 12000 g в течение 10 мин.
ПОЛУЧЕНИЕ РАДИОАКТИВНЫХ ЗОНДОВ
500 нг ДНК-матрицы и 100 нг праймера денатурировали в 10 мкл воды при 100оС в течение 10 мин и быстро охлаждали до 0оС. Затем добавляли 10 мкл 10-кратного буфера (40 мМ KP04 рН 7,5, 6,6 мМ MgCl2 и 1,0 мМ 2-меркаптоэтанол), 0,1-0,5 мКи 32Р-dATФ, 10 мкл 2 мМ раствора трех других немеченых дНTФ до концентрации 200 мкМ. Объем реакционной смеси доводили водой до 100 мкл, добавляли 3 мкл фрагмента Кленова ДНК-полимеразы I (10-12 е.а.) и инкубировали при комнатной температуре в течение 30 мин. Очистку зонда проводили методом гель - фильтрации на колонке с биогелем П-10 (Маниатис и др., 1984).
ВЫДЕЛЕНИЕ ПЛАЗМИДНОЙ ДНК
Клетки Escherichia coli, выращенные в среде Луриа-Бертани (1% NaCl, 1% бакто-триптон, 0,5% бакто-дрожжевой экстракт, рН 7,5) (среда LB) с ампициллином (50 мкг/мл), осаждали центрифугированием при 4000 g. Затем осадок суспендировали в буфере STET (8% сахароза, 0,5% тритон X-100, 10 мМ трис-HCl и 50 мМ Na2ЭДТА, рН 8,0) и добавляли лизоцим до концентрации 0,8 г/мл. Смесь инкубировали при комнатной температуре в течение 5 мин и при 100оС в течение 1 мин. После этого клеточный дебрис осаждали центрифугированием при 16000 g в течение 15 мин.
Для осаждения плазмидной ДНК к супернатанту добавляли 1/10 объема 3 М раствора ацетата натрия, равный объем изопропанола и центрифугировали при 16000 g в течение 15 мин при 20оС. Затем промывали осадок добавлением 80% этанола и повторным центрифугированием в течение 5 мин при 20оС (Маниатис и др., 1984).
ОЧИСТКА ПЛАЗМИДНОЙ ДНК МЕТОДОМ РАВНОВЕСНОГО ЦЕНТРИФУГИРОВАНИЯ В ГРАДИЕНТЕ ПЛОТНОСТИ CsCl
На каждые 10 мл раствора плазмидной ДНК, полученного после осаждения лизированных клеток (см. Выделение плазмидной ДНК), добавляли 1 г сухого CsCl, соль растворяли, после чего на каждые 10 мл раствора добавляли 0,8 мл раствора бромистого этидия с концентрацией 10 мг/мл. Смесь помещали в центрифужные пробирки Beckman, заполняли доверху раствором CsCl и бромистого этидия той же плотности и центрифугировали в роторе Ti 60 при 45000 об/мин и температуре 20оС в течение 36 часов. После этого отбирали кольцевую ковалентно замкнутую плазмидную ДНК и экстрагировали бромистый этидий из раствора до полного обесцвечивания равным объемом н-бутанола, предварительно насыщенного 1 М раствором NaCl.
ДНК осаждали центрифугированием при 15000 g, добавив равный объем 1 М раствора NH4Cl и два объема этанола. После промывания этанолом осадок ДНК растворяли в воде до концентрации 1 мкг/мкл (Маниатис и др., 1984).
ТРАНСФОРМАЦИЯ ПРОКАРИОТИЧЕСКИХ КЛЕТОК
Химическая трансформация.
Для подготовки компетентных клеток E.coli штамм XL-1 Blue MRF ' и выращивали в 100 мл 2-кратной среды Луриа (2% бакто-триптон, 1% бакто-дрожжевой экстракт, 0,1% NaCl, 0,4% глюкоза, рН 7,0) при 37оС до оптической плотности D550=0,35. Клеточную суспензию выдерживали при 0оС в течение 2 часов. Затем клетки осаждали центрифугированием при 4000 g, температуре 4оС и суспендировали в 50 мл ледяного буфера А (100 мМ CaCl2, 70 мМ MnCl2, 40 мМ NaAc, рН 5,5). После этого суспензию клеток выдерживали при 0оС в течение 30 мин, после чего клетки осаждали и суспендировали в 5 мл буфера А с 15% глицерина.
Для трансформации к 200 мкл суспензии компетентных клеток добавляли 30 нг плазмидной ДНК, растворенной в 100 мкл воды и выдерживали при 0оС в течение 30 мин. Затем инкубировали при 37оС в течение 5 мин, после чего добавляли 3 мл среды LB и инкубировали при 37оС еще 90 мин. Клетки собирали центрифугированием и высевали на селективную среду (Мазин и др., 1988).
Электротрансформация.
Для подготовки компетентных клеток E.coli штамм XL-1 Blue MRF ' выращивали в 100 мл среды LB (см. Химическая трансформация) при комнатной температуре до оптической плотности D550=0,45. Затем клетки осаждали центрифугированием при 4000 g, 4оС и промывали суспендированием последовательно в 100 мл, 50 мл и 10мл 10% раствора глицерина при 0-4оС. После этого клетки суспендировали в 240 мкл ледяного раствора GYT (10% глицерин, 0,125% бакто-дрожжевой экстракт, 0,25% бакто-триптон).
Для трансформации к 40 мкл суспензии компетентных клеток добавляли 1 мкл ДНК вектора (50 нг) при 0-4оС и пропускали электрический разряд (U=18000 В). Затем добавляли 1 мл SOC (2% бакто-триптон, 0,5% бакто-дрожжевой экстракт, 10 мМ NaCl, 2,5 мМ KCl, 10 мМ MgCl2, 10 мМ MgSO4, 20 мМ глюкоза), перемешивали, переносили в 3 мл среды SOC, прогретой до 42оС, и инкубировали при 37оС в течение 1 часа (Wai Lin Tung and King-C. Chow, 1995).
ВЫДЕЛЕНИЕ ЛЕЙКОЦИТОВ ИЗ КРОВИ РЫБ
Кровь собирали на холоду через каудальную артерию с добавлением на каждые 5 мл крови 1 мл 2,7% раствора Na2ЭДТА, рН 7,4. К выделенной крови добавляли равный объем трис-солевого буфера (TBS) (0,14 М NaCl, 5 мМ KCl, 25 мМ трис-HCl, рН 7,4). Смесь наносили при комнатной температуре на равный объем среды для выделения лейкоцитов, содержащей 24 части 9% фикола и 10 частей 34% верографина. После центрифугирования при 400 g в течение 30 мин отбирали слой лейкоцитов и разводили в большом объеме буфера TBS. После повторного центрифугирования при 300 g в течение 40 мин для лизиса остаточных эритроцитов клетки суспендировали в растворе, содержащем 9 объемов 0,83% раствора NH4Cl и 1 объем 2,06% раствора трис-HCl, рН 7,62, до концентрации 108 кл/мл и добавляли 5 объемов среды. После осаждения клеток при 300 g в течение 10 мин процедуру очистки повторили (Фримель, 1987).
ВЫДЕЛЕНИЕ СУММАРНОЙ РНК ИЗ ЛЕЙКОЦИТОВ
Осадок лейкоцитов после выделения и очистки суспендировали в 10 объемах (1 мл) раствора 1, содержащего 4 М гуанидин тиоцианат, 25 мМ цитрат натрия, рН 7,0, 0,5% саркозил и 0,1 М 2-меркаптоэтанол, при комнатной температуре. Затем добавляли 0,1 мл 2 М раствора NaAc, рН 4,0, 1 мл фенола, 0,2 мл хлороформа и перемешивали. Затем центрифугировали при 10000 g в течение 20 мин при 4оС, отбирали водную фазу, добавляли к ней равный объем изопропанола и осаждали РНК центрифугированием при 15000 g в течение 15 мин при 4оС (Chomczynski and Sacchi, 1987).
ВЫДЕЛЕНИЕ ВЫСОКОМОЛЕКУЛЯРНОЙ ДНК ИЗ ЭУКАРИОТИЧЕСКИХ КЛЕТОК
1 г печени стерляди, замороженной в жидком азоте, растирали в тонкий порошок и гомогенизировали в 15 мл раствора, содержащего 0,5 М Na2ЭДТА, рН 8,0, 0,5% саркозила, 100 мкг/мл протеиназы К и нагретого до 65оС. Затем инкубировали при 50оС течение 3 часов при легком покачивании. Затем, избегая резких встряхиваний, экстрагировали ДНК равным объемом фенола три раза, каждый раз разделяя фракции центрифугированием при 4000 g и комнатной температуре. Фенол предварительно очищали перегонкой и насыщали буфером, содержащим 1 М трис-HCl, один раз и два раза буфером, содержащим 0,1 М трис-HCl и 0,1% 2-меркаптоэтанол. Диализ ДНК проводили при 10оС в течение 12 часов против 4 л буфера, имеющего состав:
50 мМ трис-HCl, рН 8,0,
10 мМ Na2ЭДТА,
10 мМ NaCl.
Затем обрабатывали пробу РНКазой, свободной от ДНКазы,при 37оС в течение 1 часа в концентрации 100 мкг/мл. После этого экстрагировали ДНК равным объемом смеси фенол-хлороформ (1:1) два раза и проводили диализ при 10оС в течение 36 часов против 10 л буфера TE (10 мМ трис-HCl, 1 мМ Na2ЭДТА, рН 7,5), меняя буфер через каждые 12 часов (Маниатис и др., 1984).
КОНСТРУИРОВАНИЕ БИБЛИОТЕКИ кДНК
Выделение поли(A)+РНК.
Для ингибирования РНКазы все растворы, не содержащие трис, обрабатывали 0,1% диэтилпирокарбонатом 12 часов при 37оС и автоклавировали. Посуду прокаливали при 250оС в течение 4 часов. Поли(A)+РНК выделяли из суммарной РНК лейкоцитов стерляди методом хроматографии на олиго(dT)-целлюлозе. Для этого РНК растворяли в воде, прогревали при 65оС в течение 5 мин, добавляли равный объем 2-кратного буфера для нанесения РНК (40 мМ трис-HCl, рН 7,6, 1 М NaCl, 2 мМ Na2ЭДТА, 0,2% SDS) и трижды пропускали через колонку с олиго(dT)-целлюлозой. Затем промывали колонку 5-10 объемами буфера для нанесения и 4 объемами этого же буфера, содержащего 0,1 М NaCl. После этого элюировали поли(A)+РНК в 2-3 объемах воды, обработанной диэтилпирокарбонатом, добавляли 2,2 объема этанола и осаждали центрифугированием в течение 10 мин при 12000 g при 4оС (Маниатис и др., 1984).
Синтез кДНК.
Синтез кДНК проводили в соответствии с протоколом фирмы Stratagene. По матрице поли(A)+РНК синтезировали первую цепь кДНК (рис. 9).
Реакционная имела состав:
5 мкл 10-кратного буфера для синтеза первой цепи,
3 мкл смеси метил-dНTФ,
2 мкл раствора праймер-линкера (1.4 мкг/мкл),
1 мкл ингибитора РНКазной активности,
1.5 мкл фермента обратная транскриптаза (50 е.а./мкл),
вода до суммарного объема 50 мкл.
Смесь инкубировали при 37оС в течение 1 часа. Затем охлаждали до 0оС и добляли компоненты для синтеза второй цепи кДНК:
20 мкл буфера для синтеза второй цепи,
6 мкл смеси dНTФ,
88,9 мкл стерильной воды,
2 мкл 32Р-dНTФ (800 Ки/мМ),
2 мкл раствора РНКазыH (1.5 е.а./мкл),
11.1 мкл ДНК полимеразы I.
Смесь инкубировали при 16оС в течение 2,5 часов, затем охлаждали до 0оС и добавляли компоненты для застройки “липких” концов:
23 мкл смеси дНTФ,
2 мкл Pfu ДНК полимеразы (2,5 е.а./мкл).
Смесь инкубировали при 72оС в течение 30 мин.
Затем проводили экстракцию равным объемом смеси фенол-хлороформ (1:1) и равным объемом хлороформа, после чего отбирали водную фазу, добавляли 20 мкл 3М NaAc и 400 мкл 96% этанола и осаждали кДНК центрифугированием при 12000 g в течение 12 мин.
Сшивка молекул кДНК с вектором.
кДНК растворяли с EcoR I адапторами (рис. 9) и инкубировали при 4оС в течение 30 мин. Затем добавляли компоненты для сшивки молекул адапторов и молеул кДНК:
праймер-линкер
5' CTCGAGTTTTTTTTTTTT 3'
![]() 3' AAAAAAAAAAAA
5'
3' AAAAAAAAAAAA
5'
|
![]()
Реферат опубликован: 18/04/2005 (16689 прочтено)